

La lactate déshydrogénase (LDH) est une enzyme fondamentale dans le métabolisme énergétique, jouant un rôle pivot dans la glycolyse anaérobie. Son activité est intrinsèquement liée à la conversion du lactate en pyruvate, un processus essentiel à la régénération du NAD+ nécessaire à la poursuite de la glycolyse en l'absence d'oxygène. Cette enzyme, présente dans une vaste gamme d'organismes, des bactéries aux mammifères, est d'un grand intérêt pour la recherche biochimique et médicale. Ce compte rendu détaille les étapes d'une procédure de purification de la LDH à partir d'un extrait de tissu bovin fœtal, en mettant l'accent sur la technique de chromatographie d'affinité et les méthodes de quantification de l'activité enzymatique et de la concentration protéique.
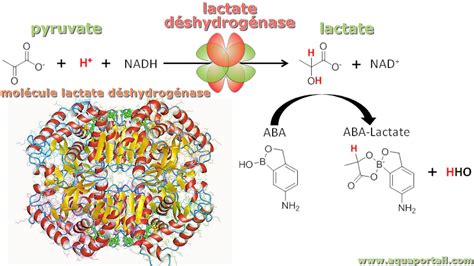
La lactate déshydrogénase catalyse la réaction réversible suivante :
L-Lactate + NAD+ ⇌ Pyruvate + NADH + H+
Cette réaction est au cœur de la fermentation lactique, un processus qui se déroule dans les organismes en conditions d'anaérobiose. Le rôle principal de la LDH dans ce contexte est de régénérer le NAD+ à partir du NADH, permettant ainsi à la glycolyse de se poursuivre. Le NADH, Nicotinamide Adénine Dinucléotide sous sa forme réduite, agit comme cofacteur dans cette réaction, étant converti en NAD+ lors de la transformation du pyruvate en L-lactate.
Chez les mammifères, la LDH est un tétramère composé de quatre sous-unités, qui peuvent être de deux types : M (pour "muscle") et H (pour "heart"). Cette combinaison donne naissance à cinq isoenzymes tétramériques distinctes : M4, M3H, M2H2, MH3 et H4. La prédominance de la sous-unité H est observée dans le muscle cardiaque, tandis que la sous-unité M est plus abondante dans les tissus comme le muscle squelettique ou le foie. Les différentes isoenzymes présentent des variations dans leurs constantes cinétiques et leurs pH isoélectriques, ces derniers permettant leur séparation par électrophorèse. Par exemple, le monomère H est plus chargé négativement à pH neutre ou alcalin que le monomère M.
Le but principal de cette manipulation est d'extraire et de purifier la lactate déshydrogénase (LDH) présente dans un extrait de tissu bovin fœtal. L'objectif secondaire est de quantifier l'activité enzymatique de la LDH à différentes étapes de la purification, ainsi que de déterminer les concentrations protéiques afin d'en déduire l'enrichissement et le rendement de l'enzyme.
La purification de la LDH a été réalisée par chromatographie d'affinité, une technique chromatographique particulièrement efficace pour séparer les composants d'un mélange. Le principe fondamental de cette méthode repose sur l'affinité spécifique qu'une molécule présente pour un ligand immobilisé sur une phase stationnaire. Cette technique permet potentiellement de purifier une molécule d'intérêt en une seule étape, ce qui en fait une méthode de choix pour l'isolement d'enzymes.
La chromatographie d'affinité implique l'utilisation d'une colonne remplie d'une matrice insoluble (phase stationnaire) à laquelle est attaché un ligand spécifique de la molécule à purifier. Le mélange contenant la molécule d'intérêt est introduit dans la colonne. La molécule cible se lie de manière réversible au ligand, tandis que les autres composants du mélange sont éliminés par un lavage de la colonne. L'étape finale, appelée élution, consiste à détacher la molécule purifiée du ligand en utilisant une phase mobile appropriée, permettant ainsi sa récupération.
Pour une chromatographie d'affinité réussie, il est crucial de connaître la structure et la spécificité de la molécule à purifier afin de sélectionner un ligand approprié. Ce ligand doit avoir une affinité forte et sélective pour la molécule d'intérêt, tout en minimisant les interactions non spécifiques avec d'autres composés.
Dans le cas de la LDH, son utilisation du NADH comme cofacteur suggère que le NADH peut servir de ligand. Cependant, pour cette purification, une molécule appelée "bleu de Cibacron" a été utilisée. Le bleu de Cibacron est un colorant qui présente une analogie structurelle avec le cofacteur NAD+. Cette molécule analogue est fixée par liaison covalente sur des billes de gel d'agarose, qui constituent la phase stationnaire. L'ensemble du processus de chromatographie d'affinité se déroule en quatre étapes clés : l'équilibration, la fixation, le rinçage et l'élution.
L'étape d'équilibration a consisté à placer le gel de bleu de Cibacron dans une mini-colonne et à l'équilibrer avec une solution tampon de lavage (PiNa, pH 7). Le but de cette étape est double : premièrement, éliminer l'éthanol présent dans le gel d'agarose, utilisé comme agent germicide. L'éthanol est susceptible de dénaturer les protéines, y compris la LDH, ce qui rendrait la purification inefficace. Deuxièmement, cette étape permet d'établir un pH neutre, une condition chimique généralement favorable à la fixation des enzymes sur leur ligand.
Durant l'étape de fixation, l'extrait de tissu fœtal bovin contenant la LDH est introduit dans la mini-colonne. La lactate déshydrogénase présente dans l'extrait se lie spécifiquement au gel de bleu de Cibacron. La fraction du lysat qui ne se lie pas au gel s'écoule hors de la colonne et est récupérée. Cette fraction est appelée "fraction non retenue".
Suite à la fixation, un rinçage de la colonne est effectué avec la solution tampon de lavage. Cette étape vise à éliminer de manière exhaustive les composants de l'extrait qui n'ont pas interagi avec le gel de bleu de Cibacron, ainsi que les liaisons non spécifiques. La fraction issue de ce rinçage est également conservée, car elle pourrait contenir des protéines faiblement liées.
L'étape finale est l'élution de la LDH purifiée. Pour ce faire, une solution tampon contenant du NADH et de l'acide oxamique est utilisée. Le NADH est le cofacteur naturel de la LDH, tandis que l'acide oxamique est un inhibiteur compétitif analogue du pyruvate, le substrat de la réaction. Ces deux molécules, le NADH et l'acide oxamique, ont une affinité pour la LDH qui est plus forte que celle du bleu de Cibacron. En conséquence, elles entrent en compétition avec le bleu de Cibacron pour la liaison à l'enzyme, provoquant ainsi le détachement de la lactate déshydrogénase de la phase stationnaire. L'élution est généralement réalisée en plusieurs fractions pour maximiser la récupération de l'enzyme. Dans ce protocole, trois éluats (E1, E2, E3) ont été collectés, chacun correspondant à 1 mL de tampon d'élution.
Pour évaluer l'efficacité de la purification et confirmer la présence de la LDH dans les fractions éluées, l'activité enzymatique de différentes fractions (extrait brut, fraction non retenue, fraction de lavage, et éluats E1, E2, E3) a été mesurée à l'aide d'un spectrophotomètre couplé à un traceur.
Le spectrophotomètre est un appareil qui mesure l'absorbance d'une solution à une longueur d'onde spécifique. Conformément à la loi de Beer-Lambert, l'absorbance est directement proportionnelle à la concentration de la substance absorbante dans la solution. Pour la mesure de l'activité de la LDH, la longueur d'onde de 340 nm est utilisée, car c'est la longueur d'onde à laquelle le NADH absorbe fortement la lumière. Le traceur enregistre les variations d'absorbance au fil du temps. Dans la réaction catalysée par la LDH, la production de NADH entraîne une augmentation de l'absorbance à 340 nm. En suivant cette augmentation, il est possible de quantifier la vitesse de la réaction enzymatique et, par conséquent, l'activité de la LDH.
Le protocole de mesure de l'activité enzymatique implique le mélange de la fraction enzymatique à analyser avec un tampon contenant le substrat (L-lactate) et le cofacteur (NAD+) en excès. Cet excès de réactifs force la réaction à se dérouler dans le sens de la production de NADH. Les variations d'absorbance à 340 nm sont ensuite enregistrées sur une période donnée.
Les données brutes obtenues lors de la mesure de l'activité enzymatique sont généralement des variations d'absorbance par unité de temps (ΔA/Δt). Ces valeurs sont ensuite converties en vitesse enzymatique (Vi) en utilisant la loi de Beer-Lambert :
Vi = (ΔA/Δt) × (1 / (ε × l)) × Facteur de conversion
où :
Suite à ce calcul, les vitesses mesurées pour un volume de 50 µL de fraction sont multipliées par 20 pour obtenir l'activité totale enzymatique par mL.
Les valeurs d'activité enzymatique obtenues pour les différentes fractions sont les suivantes (en µmoles/minute/mL) :
Ces résultats sont cohérents avec les attentes. L'extrait brut présente l'activité enzymatique la plus élevée, ce qui est normal puisqu'il contient l'enzyme dans sa concentration native. On observe une diminution significative de l'activité dans la fraction non retenue, indiquant que la majeure partie de la LDH s'est fixée au gel. La fraction de rinçage ne présente pratiquement aucune activité, ce qui confirme que les impuretés non liées ont été efficacement éliminées. Les éluats E1, E2 et E3 montrent une activité enzymatique détectable, avec une décroissance progressive de l'activité au fil des éluats, ce qui est attendu lors d'une élution fractionnée. La présence d'une pente non nulle pour la fraction non retenue suggère qu'une petite quantité de LDH n'a pas été retenue par la colonne, mais cette valeur est très faible. La quasi-absence de LDH dans la fraction de rinçage atteste de l'efficacité de la manipulation sur la colonne d'élution pour la purification de l'enzyme.
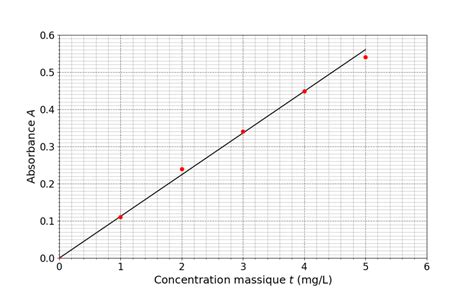
Pour calculer l'enrichissement et le rendement de la LDH, il est nécessaire de connaître la concentration totale de protéines dans chaque fraction. La méthode de Bradford est couramment utilisée à cette fin. Elle repose sur la réaction du réactif de Bradford avec les protéines, entraînant un changement de couleur proportionnel à la concentration protéique. L'absorbance de la solution est mesurée à 595 nm à l'aide d'un spectrophotomètre.
Pour ce faire, une gamme étalon est réalisée en préparant des solutions contenant des quantités croissantes de BSA (Albumine Sérique Bovine), une protéine standard. Ces solutions, mélangées au réactif de Bradford, permettent de construire une courbe d'étalonnage reliant l'absorbance à la concentration massique en protéines. Les absorbances des extraits brut et des fractions purifiées sont ensuite mesurées dans les mêmes conditions. En reportant ces valeurs sur la courbe d'étalonnage, les concentrations massiques en protéines (en µg/mL) des différents extraits peuvent être déterminées.

Une fois la concentration massique en protéines connue pour chaque fraction, l'activité spécifique de la LDH peut être calculée. L'activité spécifique est définie comme le rapport entre l'activité enzymatique totale de la LDH dans un extrait et la quantité totale de protéines dans cet extrait. Une activité spécifique élevée indique une plus grande pureté de l'extrait, c'est-à-dire une proportion plus importante de LDH par rapport aux autres protéines. L'activité spécifique est généralement exprimée en µmoles/min/mg de protéine.
L'enrichissement de la fraction E1 par rapport à l'extrait brut est calculé comme suit :
Enrichissement E1 = Activité Spécifique E1 / Activité Spécifique Extrait Brut
Le rendement total de l'activité enzymatique dans l'extrait brut est un indicateur de la quantité d'enzyme initialement présente. Le rendement de la purification est ensuite calculé en comparant l'activité enzymatique totale de l'extrait purifié à celle de l'extrait brut. Un rendement élevé signifie qu'une grande partie de l'activité enzymatique initiale a été conservée au cours du processus de purification. Le rendement total de l'activité enzymatique dans l'extrait brut, tel que mentionné dans les données fournies, s'élève à 69.65% de l'activité enzymatique totale.
La purification par chromatographie d'affinité sur bleu de Cibacron s'est avérée efficace pour isoler la lactate déshydrogénase de l'extrait de tissu bovin fœtal. Les données d'activité enzymatique montrent une concentration significative de LDH dans les fractions éluées (E1, E2, E3), avec une diminution notable dans les fractions non retenue et de lavage. Cette observation confirme que la majorité de l'enzyme a été retenue par la colonne et récupérée lors de l'élution.
L'activité spécifique calculée pour chaque fraction permet d'évaluer la pureté de la LDH. Une augmentation de l'activité spécifique au cours des étapes de purification est un indicateur direct du succès de la séparation des protéines indésirables. L'enrichissement de la fraction E1, calculé par rapport à l'activité spécifique de l'extrait brut, quantifie l'augmentation de la concentration relative de LDH.
Le rendement global de la purification, bien qu'il ne soit pas explicitement calculé dans les données fournies pour les fractions finales, est un paramètre crucial. Un rendement de 69.65% pour l'activité enzymatique totale dans l'extrait brut indique la quantité initiale d'enzyme disponible. La perte d'activité au cours de la purification est inévitable et peut résulter de divers facteurs, tels que des liaisons non optimales au ligand, une élution incomplète, ou une dénaturation partielle de l'enzyme.
Il est important de noter que le bleu de Cibacron est un ligand relativement peu spécifique, ce qui signifie qu'il peut se lier à d'autres protéines possédant une structure analogue aux nucléotides. Par conséquent, la purification obtenue avec cette méthode peut être considérée comme partielle, et les fractions éluées peuvent encore contenir une certaine quantité d'impuretés protéiques. Pour obtenir une protéine hautement pure, des étapes de purification supplémentaires, telles qu'une chromatographie d'échange d'ions ou de filtration sur gel, pourraient être nécessaires.
L'étude de la lactate déshydrogénase et de ses isoenzymes a des implications importantes dans le domaine médical. Par exemple, l'augmentation du taux d'isoenzyme H4 de la LDH dans le sérum est un marqueur diagnostique de l'infarctus du myocarde. La capacité à purifier et caractériser ces enzymes est donc essentielle pour la recherche fondamentale et le développement de méthodes diagnostiques et thérapeutiques.
Les recherches sur des organismes tels que l'escargot Otala lactea ont montré que les modifications post-traductionnelles (PTMs) peuvent influencer l'activité de la LDH, notamment en réponse à des conditions de stress. Par exemple, la phosphorylation de la LDH du muscle de la patte chez cet escargot en estivation conduit à une meilleure efficacité dans la consommation du pyruvate et à une stabilité thermique accrue, mais une stabilité structurelle réduite. De même, la déphosphorylation de la LDH de l'hépatopancréas en estivation diminue son efficacité dans la réaction directe, mais améliore la tolérance au sel lorsque l'eau devient rare. Ces découvertes soulignent la complexité de la régulation enzymatique et son adaptation aux conditions environnementales.
En conclusion, la purification de la lactate déshydrogénase par chromatographie d'affinité sur bleu de Cibacron est une méthode efficace pour isoler cette enzyme d'un extrait tissulaire. Les mesures d'activité enzymatique et de concentration protéique permettent d'évaluer le succès de la purification en termes d'enrichissement et de rendement, tout en fournissant des données précieuses pour la compréhension de la biochimie de cette enzyme clé.
tags: #tp #lactate #deshydrogenase #purification #compte #rendu
