

L'identification précise et rapide des agents infectieux est une priorité absolue, particulièrement lors d'une épidémie. Si les méthodes phénotypiques ont longtemps dominé les laboratoires de microbiologie clinique, l'avènement de la biologie moléculaire a marqué un tournant décisif. Désormais, l'identification des pathogènes s'appuie sur la comparaison de leur "empreinte moléculaire" avec des bases de données exhaustives. Cependant, le séquençage haut débit promet d'aller au-delà de cette simple identification, en exploitant la totalité des informations contenues dans le génome d'un pathogène. Cette transition, passant de l'empreinte au "portrait-robot" génomique, soutenue par un nombre croissant d'études préliminaires, ouvre la voie à une adaptation plus fine et plus ciblée des traitements aux spécificités de chaque agent infectieux.
La notion de séquençage haut débit a émergé au début des années 2000 avec la première génération de machines, aujourd'hui largement oubliée, proposées par Lynx Therapeutics. Cinq ans plus tard, l'arrivée des séquenceurs de deuxième génération a initié une ère de séquençage multi-parallélisé, dont les applications dans les laboratoires de recherche n'ont cessé de croître. Depuis, le nombre de génomes entièrement séquencés et accessibles dans les bases de données a explosé de manière exponentielle. En 2014, deux générations de séquenceurs coexistent : les séquenceurs de deuxième génération, dont certains sont déclinés en versions de paillasse offrant des débits généralement suffisants pour les applications en microbiologie, et les séquenceurs de troisième génération.
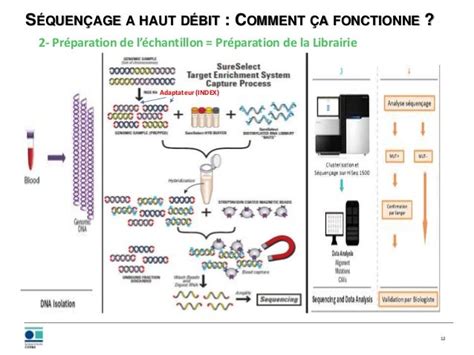
Le principe fondamental du séquençage haut débit repose sur la parallélisation des réactions, permettant le séquençage de courtes lectures d'une librairie (reads). Les applications liées à cette technologie impliquent une phase de "biologie humide" et une phase de "biologie sèche" (Figure 1).
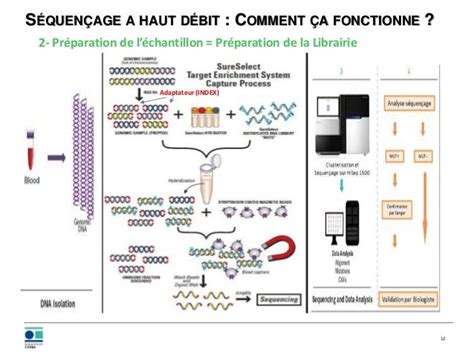
En laboratoire de biologie moléculaire, la phase de biologie humide débute par l'extraction et la purification de l'ADN du pathogène à séquencer. Le pathogène peut être isolé préalablement ou non, cette modalité ayant un impact principal sur la puissance de séquençage nécessaire et les différents types d'analyses bioinformatiques à réaliser. Les modes d'amplification clonale peuvent varier selon les fournisseurs de séquenceurs, et font aujourd'hui l'objet d'une simplification des procédures grâce à des automates dédiés. Les technologies de séquençage se distinguent principalement par leurs modes de détection, qui produisent un signal fonction des bases polymérisées lors de l'opération de séquençage multi-parallélisé. La phase de biologie sèche vise à générer de l'information biologique pertinente à partir des reads, par exemple l'identification de gènes présents ou de variations par rapport à une référence, à l'aide d'outils bioinformatiques.
Afin d'obtenir des séquences plus longues à partir des reads, deux approches principales peuvent être employées : le mapping sur référence et l'assemblage de novo.
Le mapping sur référence consiste à repositionner les reads sur un génome de référence. Cette référence est le plus souvent constituée par le génome de l'organisme séquencé ou par un génome proche, complètement annoté et disponible dans les bases de données publiques. Dans le cas d'un séquençage à partir d'un prélèvement, la référence n'est pas connue a priori. Cependant, il est possible d'identifier, à l'issue du séquençage, un ou plusieurs génomes qui serviront de références en exploitant l'homologie entre les reads et les génomes disponibles dans les banques publiques. Le mapping sur référence permet, par comparaison, de reconstruire la séquence génomique de l'organisme séquencé et d'identifier les différences par rapport au génome de référence.
Lorsque aucun génome proche de celui de l'organisme étudié n'est disponible dans les banques de données, l'assemblage de novo est utilisé. Cette méthode permet de construire, sans a priori, des séquences plus longues (contigs) à partir des reads.
Au cœur de nombreuses technologies de séquençage de nouvelle génération se trouvent les nucléotides fluorescents terminateurs réversibles (F-NRTs). Ces molécules jouent un rôle essentiel en permettant l'arrêt contrôlé de la polymérisation de l'ADN, tout en portant un marqueur fluorescent qui révèle l'identité de la base incorporée. Le caractère "réversible" du terminateur est la clé de la capacité à poursuivre le séquençage après l'identification de chaque base.
La méthode de Sanger, bien que fondamentale, reposait sur des ddNTPs (didésoxyribonucléotides) qui, une fois incorporés, bloquaient définitivement la chaîne d'ADN. Les F-NRTs, en revanche, possèdent une modification chimique au niveau du groupe 3'-OH, qui bloque temporairement la polymérisation. Après détection de la fluorescence, ce groupe bloqueur peut être clivé chimiquement, restaurant ainsi le groupe 3'-OH et permettant la poursuite de la réaction de synthèse.
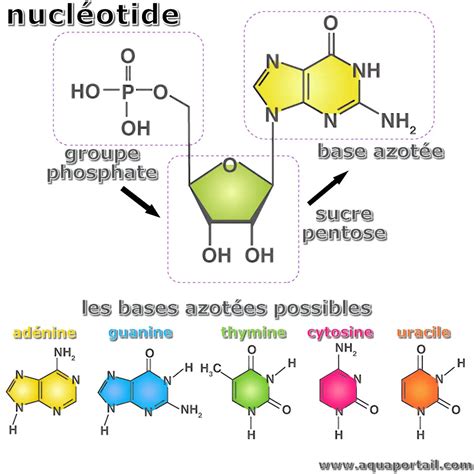
Ces nucléotides modifiés portent généralement un groupe protecteur au niveau du 3'-OH, tel qu'un groupe 3'-O-méthylazido ou 3'-O-(2-nitrobenzyl). Ces groupes sont conçus pour être chimiquement clivables. La fluorescence est quant à elle attachée à la base nucléique via un linker, souvent un groupe 2-nitrobenzyl. La spécificité et la réversibilité de ces modifications sont cruciales pour la précision et l'efficacité du séquençage.
Parmi les exemples de F-NRTs décrits dans la littérature scientifique, on trouve des analogues tels que le ddCTP-PC-Bodipy-FL-510, le ddUTP-PC-R6G, le ddATP-PC-ROX et le ddGTP-PC-Bodipy-650. Ces molécules, lorsqu'elles sont incorporées dans une chaîne d'ADN en cours de synthèse, arrêtent temporairement la polymérisation. L'irradiation subséquente avec une lumière d'une longueur d'onde spécifique (par exemple, 355 nm pour les groupes 2-nitrobenzyl) entraîne le clivage du groupe photolabile, libérant ainsi le fluorophore et restaurant le groupe 3'-OH.
Une autre approche utilise des groupes 3'-O-azido qui peuvent être réduits par des réactifs tels que le Tris(2-carboxy-éthyl) phosphine (TCEP) pour régénérer le groupe 3'-OH. Ces nucléotides, appelés 3'-O-N3-dNTPs, sont ensuite utilisés en combinaison avec des ddNTPs marqués par fluorescence. La réaction de Staudinger, impliquant le TCEP, permet de cliver le groupe azido-méthyl et de restaurer le groupe hydroxyle, permettant ainsi la poursuite de l'élongation de la chaîne d'ADN.
L'avantage majeur de l'utilisation de F-NRTs réside dans la possibilité de réaliser un séquençage par synthèse (SBS - Sequencing by Synthesis) en mode "quatre couleurs". Cela signifie que les quatre types de nucléotides (A, T, C, G) peuvent être ajoutés simultanément dans le mélange réactionnel, chacun portant un fluorophore distinct. Après l'incorporation, le signal fluorescent émis par le nucléotide ajouté est détecté. Ce signal révèle l'identité de la base incorporée à cette position. Ensuite, le terminateur réversible et le fluorophore sont clivés, permettant à la polymérase d'ajouter le nucléotide suivant. Ce cycle de synthèse, détection et clivage est répété jusqu'à l'obtention de la séquence désirée.
Les laboratoires de microbiologie clinique ont pour mission de fournir rapidement des éléments de caractérisation des microorganismes responsables de maladies infectieuses. Si l'identification est souvent réalisée après isolement par des méthodes phénotypiques, les méthodes de biologie moléculaire, comme la PCR en temps réel, permettent une identification sans isolement préalable. Ces méthodes sont sensibles, standardisables et relativement faciles à mettre en œuvre.
Cependant, l'approche réductrice basée sur les empreintes moléculaires présente des limites, notamment en ignorant la plasticité des génomes microbiens, comme le rôle des intégrons dans la résistance aux antibiotiques. Le séquençage haut débit, en palliant ces défauts, offre la possibilité d'obtenir une connaissance quasi exhaustive du génome du pathogène. Cela permet de réaliser des "antibiogrammes in silico", d'estimer le pouvoir pathogène d'une souche et, in fine, de proposer un traitement ciblé.
Le séquençage haut débit permet de caractériser des génomes complets associés à une infection, ouvrant la voie à une évolution des pratiques hospitalières. Au-delà des informations fournies par l'antibiogramme, la connaissance des communautés de gènes impliqués dans une infection particulière pourrait mener à de nouvelles approches thérapeutiques plus ciblées et potentiellement plus efficaces.
Pour une utilisation en routine, la tendance est à la simplification des procédures techniques, dont le temps de réalisation se réduit à quelques heures. Le séquenceur délivre des données brutes, qu'il faut reconstituer à l'aide d'outils bioinformatiques. L'exploitation rapide, rationnelle et automatisée des données est un enjeu majeur de la phase de bioanalyse. Les défis actuels résident principalement dans la partie analytique : les outils disponibles sont souvent difficiles d'accès pour les non-experts, nécessitant des compétences informatiques avancées et des interfaces graphiques intuitives. Une réflexion sur la centralisation des données au sein de banques dédiées et sur la standardisation des formats est également nécessaire pour regrouper efficacement les données pertinentes et améliorer les connaissances.
Des difficultés persistent quant à la généralisation du séquençage haut débit pour l'élucidation des maladies infectieuses, excluant pour l'instant une utilisation en routine généralisée. Néanmoins, de nombreuses études démontrent la faisabilité et la pertinence de cette approche. Par exemple, l'étude de Sherry et al. (2013) a montré qu'il était possible d'obtenir et d'analyser des données de séquençage d'E. coli multirésistantes en cinq jours, pour un budget de réactifs de 300 US$ par souche.
La perspective d'un séquençage sans isolement préalable du pathogène permettrait de réduire encore le temps d'analyse et de s'affranchir des étapes de culture. Des approches telles que le séquençage haut débit du génome de cellules isolées par immunocapture, ou les approches métagénomiques, sont envisagées. La métagénomique vise à séquencer tous les ADN microbiens présents dans un échantillon afin de les identifier et de les caractériser. Bien que coûteuse et complexe à implémenter, elle permet de connaître la communauté de gènes impliqués dans une pathologie, d'éviter la contrainte de la mise en culture et de s'affranchir des stratégies de biologie moléculaire avec a priori. L'étude de Loman et al. (2011) a ainsi permis d'identifier et de caractériser par séquençage direct, à partir d'échantillons complexes, les souches impliquées dans un épisode infectieux à Escherichia coli entérohémorragique, y compris des souches non détectées par les approches conventionnelles.
Avec ou sans isolement préalable, le séquençage haut débit apparaît comme une approche applicable en routine dans les laboratoires de microbiologie. Les séquenceurs de paillasse, conçus pour séquencer des génomes microbiens en quelques heures, offrent une capacité suffisante et représentent un investissement acceptable. Actuellement, entre 48 et 72 heures sont nécessaires pour obtenir les reads.
Le séquençage haut débit peut permettre le décodage intégral d'un génome pathogène impliqué dans un épisode infectieux, des épidémies ou des attaques bioterroristes. L'homogénéité du type de données (séquences ADN) facilite leur exploitation par un grand nombre d'experts. L'épidémie allemande à E. coli O104:H4 de 2011 est un exemple notable de mise en commun de données de séquençage exploitées par une large communauté scientifique. Une analyse bioinformatique fine du génome complet permet d'acquérir de nouvelles connaissances sur l'organisme étudié et de compléter les bases de données utilisées initialement. Le développement d'outils bioinformatiques spécifiques et standardisés peut être mené durant cette phase et réutilisé lors des urgences. Les connaissances acquises sur les facteurs de virulence et les gènes de résistance peuvent être exploitées par les industries de la santé.
En conclusion, les nucléotides fluorescents terminateurs réversibles sont au cœur d'une technologie de séquençage qui révolutionne la microbiologie clinique. Ils permettent une approche plus complète et plus rapide de l'identification et de la caractérisation des pathogènes, ouvrant la voie à des diagnostics plus précis et à des traitements plus ciblés, contribuant ainsi à une meilleure prise en charge des maladies infectieuses.
tags: #nucleotides #fluorescentterminateurs #reversibles
