

La thermodynamique moléculaire, branche fondamentale de la thermodynamique, transcende les observations macroscopiques pour plonger au cœur des interactions à l'échelle des composants moléculaires. Elle se consacre à la compréhension et à la prédiction des propriétés thermodynamiques des matériaux en s'appuyant sur leur structure et les forces qui régissent leurs interactions moléculaires. Cette discipline est d'une importance capitale pour l'ingénierie chimique, la conception de processus optimisés, la dissection des systèmes biologiques complexes et l'innovation dans le domaine des matériaux.
Les principes de la thermodynamique du génie moléculaire établissent les bases de l'application des concepts thermodynamiques au niveau microscopique. Cette perspective permet de quantifier le comportement des molécules, que ce soit lors de leurs interactions, de leurs changements d'état ou de leur participation à des réactions chimiques. L'analyse se concentre sur les variations d'énergie, la stabilité intrinsèque et le comportement des différentes phases, le tout interprété à travers le prisme de la structure et des forces moléculaires.
Les éléments centraux de cette approche incluent :
La thermodynamique moléculaire s'articule autour d'un ensemble de principes fondamentaux qui éclairent notre compréhension des phénomènes à l'échelle moléculaire. Son champ d'application s'étend à des domaines critiques tels que l'ingénierie, la science des matériaux, la biochimie et les sciences de l'environnement. Les principes abordent les variations d'entropie et d'enthalpie, les équilibres chimiques, et la relation intrinsèque entre la structure moléculaire et les propriétés thermodynamiques observables.
Les applications de la thermodynamique moléculaire sont vastes et touchent à des aspects essentiels de la science et de la technologie :
L'étude de l'équilibre des phases fluides par la thermodynamique moléculaire se concentre sur la distribution des molécules entre différentes phases en conditions d'équilibre. Ce domaine est fondamental pour appréhender la manière dont les substances se séparent, se mélangent ou réagissent au sein des phases liquide et gazeuse. Il s'appuie sur des modèles théoriques et des simulations pour prédire comment les interactions moléculaires dictent le comportement des phases, la solubilité des composants et les phénomènes de transitions de phase.
Les concepts clés incluent :
Ces connaissances sont indispensables pour des procédés d'ingénierie chimique tels que la distillation, l'absorption et l'extraction liquide-liquide, où le contrôle des phases est primordial.
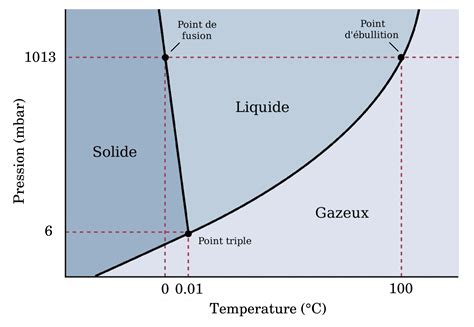
La thermodynamique moléculaire statistique joue un rôle pivot en comblant le fossé entre les observations macroscopiques et le comportement microscopique des molécules. Elle emploie des méthodes statistiques pour prédire et expliquer les propriétés thermodynamiques des systèmes en se basant sur le comportement statistique de leurs constituants moléculaires.
Les approches statistiques en thermodynamique moléculaire font appel à la mécanique statistique pour dériver les propriétés thermodynamiques à partir des caractéristiques moléculaires. Cela implique de déterminer la distribution des énergies au sein des populations moléculaires, de prédire les transitions de phase et de calculer des propriétés macroscopiques comme la pression, la température et le volume à partir des données moléculaires.
Au cœur de cette méthodologie se trouve la distribution de Boltzmann.
Distribution de Boltzmann : Cette distribution statistique fondamentale décrit la probabilité qu'un système se trouve dans un état d'énergie donné à une température spécifique. Elle est essentielle pour comprendre comment l'énergie se répartit entre les molécules d'un système en équilibre.
La thermodynamique moléculaire statistique est un outil inestimable dans de nombreuses disciplines scientifiques. Elle a révolutionné notre compréhension dans des domaines tels que la science des matériaux, où elle aide à prédire les propriétés des matériaux, et la biochimie, où elle élucide la thermodynamique du repliement et de la stabilité des protéines.
En pharmacologie, par exemple, elle est utilisée pour anticiper l'interaction entre les médicaments et leurs cibles moléculaires. Ces prédictions sont cruciales pour la conception rationnelle de nouveaux médicaments aux profils d'efficacité et de sécurité optimaux. La polyvalence de cette approche découle de sa capacité à modéliser des systèmes complexes impliquant un grand nombre de composants en interaction.
Les méthodes statistiques offrent un cadre quantitatif rigoureux pour décrire les systèmes moléculaires. Elles permettent aux scientifiques de déduire les propriétés thermodynamiques macroscopiques à partir des états microscopiques. Cela inclut la prédiction de l'impact des changements au niveau moléculaire, tels que les interactions intermoléculaires et les distributions d'énergie, sur les propriétés observables comme l'entropie, l'enthalpie et la capacité calorifique.
Une technique couramment employée est la simulation de Monte Carlo.
Simulations de Monte Carlo : Il s'agit d'une technique informatique qui utilise l'échantillonnage aléatoire pour estimer des fonctions mathématiques et simuler le comportement de divers systèmes physiques et mathématiques. Elle est particulièrement efficace pour étudier les systèmes présentant un grand nombre de degrés de liberté couplés, typiques en thermodynamique moléculaire.
L'étude de la thermodynamique moléculaire appliquée aux systèmes complexes révèle la danse subtile de l'énergie et des molécules qui sous-tend la fonction et le comportement de ces systèmes. En disséquant les interactions moléculaires et les transformations énergétiques, ce domaine améliore notre compréhension des processus chimiques et biologiques complexes.
L'analyse des systèmes complexes nécessite une approche multidimensionnelle. Elle implique l'utilisation de la mécanique statistique pour décortiquer le comportement collectif des molécules, le recours à des modèles informatiques pour simuler leur dynamique, et l'application des théories thermodynamiques pour prédire le comportement global du système. Ces analyses permettent de comprendre, au niveau moléculaire, des phénomènes tels que les changements de phase, la cinétique des réactions et le transfert d'énergie au sein de systèmes complexes.
La mécanique statistique et les modèles informatiques sont centraux dans la prédiction et la compréhension des propriétés thermodynamiques des systèmes complexes à l'échelle moléculaire. Il est essentiel de saisir comment les distributions d'énergie entre les molécules dictent les propriétés macroscopiques observables. L'entropie, par exemple, mesure le degré de désordre et joue un rôle crucial dans la détermination de la direction du flux d'énergie au sein d'un système.
La thermodynamique moléculaire est la pierre angulaire de la compréhension du fonctionnement des systèmes complexes, qu'il s'agisse des voies biochimiques complexes ou de l'efficacité des processus de conversion d'énergie. En élucidant les bases moléculaires des transformations et des interactions énergétiques, ce domaine contribue à la conception de processus chimiques plus efficaces, à la découverte de nouveaux matériaux et à la compréhension des mécanismes biologiques.
Les applications principales incluent :
Systèmes complexes : Ce sont des systèmes composés d'éléments interconnectés dont le comportement global présente des propriétés qui ne sont pas évidentes à partir des propriétés de leurs composants individuels.
La thermodynamique moléculaire fournit les fondements théoriques nécessaires pour manipuler et contrôler des systèmes complexes au bénéfice de l'humanité, en faisant le lien entre les phénomènes moléculaires et leurs applications concrètes.
La thermodynamique moléculaire des solutions électrolytiques se penche sur les propriétés et comportements thermodynamiques spécifiques des solutions contenant des ions. Ce domaine fusionne les principes de la chimie physique et de la thermodynamique pour comprendre comment les électrolytes influencent des phénomènes tels que la solubilité, le comportement des phases et la conductivité électrique.
Les principes fondamentaux de la thermodynamique moléculaire dans les solutions d'électrolytes posent les bases de la compréhension des caractéristiques distinctives de ces systèmes. Au cœur de cette étude se trouvent les interactions entre les ions de l'électrolyte et les molécules du solvant, et comment cette relation affecte les propriétés globales de la solution.
Les concepts clés incluent :
La théorie de Debye-Hückel, par exemple, est essentielle pour comprendre les effets de la concentration de l'électrolyte sur des propriétés telles que la force ionique et les coefficients d'activité, ce qui est crucial pour la conception de processus impliquant ces solutions.
Théorie de Debye-Hückel : Ce modèle mathématique décrit la distribution des charges électriques autour d'un ion dans une solution, aidant à comprendre les interactions ioniques. C'est une pierre angulaire pour prédire le comportement des solutions ioniques dans diverses conditions.
Les principes de la thermodynamique moléculaire trouvent de nombreuses applications dans les solutions électrolytiques, impactant significativement diverses industries et domaines de recherche. Ces applications comprennent le développement d'électrolytes pour les batteries et les piles à combustible, la compréhension des processus de corrosion, la conception de membranes pour la séparation ionique, et l'étude des solutions salines dans les océans et les systèmes biologiques.
Le laboratoire TIM, par exemple, combine la thermodynamique expérimentale avec l'interprétation par simulation moléculaire pour étudier les relations structure-propriétés des fluides et des interfaces. Ils se concentrent sur le lien entre les interactions et structures moléculaires, abordées d'un point de vue fondamental, et les grandeurs macroscopiques nécessaires au développement de nouveaux matériaux, dispositifs ou procédés. Les thématiques de recherche s'articulent autour des Fluides pour la Production et l'Extraction d'Énergie (FEE) et des Interfaces Moléculaires et Modélisation Multi-échelles (I3M) fluides-matériaux.
La thermodynamique moléculaire est essentiellement l'application de la mécanique statistique au calcul des grandeurs thermodynamiques et de transport des fluides ou des matériaux. Par rapport à la thermodynamique classique, qui est générale et macroscopique, elle part d'une connaissance des structures et des interactions à l'échelle moléculaire pour atteindre des grandeurs importantes pour la chimie, l'ingénierie, les sciences de l'environnement et du vivant. Les grandes évolutions des dernières années ont été la montée en puissance de la chimie quantique, qui est la base pour décrire les interactions moléculaires, et de la simulation moléculaire, qui permet d'étudier le comportement collectif de plusieurs milliers de particules.
Apparue au milieu du XXe siècle, la simulation moléculaire est aujourd'hui un outil largement utilisé pour aider à interpréter et comprendre des résultats expérimentaux, tester de nouvelles théories, ou prédire le comportement physique ou chimique de la matière. Ludwig Eduard Boltzmann n’a pas pu utiliser la puissance des ordinateurs modernes, mais ce père de la thermodynamique statistique, fervent « atomiste », a permis aux chimistes théoriciens d’aujourd’hui de réaliser leurs propres expériences ! Elles ne sont certes ni « in vivo » ni « in vitro », mais la modélisation moléculaire est véritablement une expérience « in silico ».
Dans la deuxième moitié du XIXe siècle, la communauté scientifique internationale adopte progressivement l’idée selon laquelle la matière est faite d’atomes et de molécules. Un double défi est alors à relever. Il s’agit d’une part de développer des modèles de la matière à l’échelle microscopique (ce qui conduira au développement de la mécanique quantique), et d’autre part de faire ensuite le lien entre l’échelle atomique et l’échelle macroscopique, ce qui conduira à la thermodynamique statistique. Au premier de ces défis sont associés les noms de Bohr, Schrödinger, Heisenberg, de Broglie… Au second, ceux de Maxwell, Boltzmann et Gibbs. Au moment où se produisent ces grandes évolutions scientifiques (au début du XXe siècle), le rêve est d’aboutir à une explication complète du monde fondée sur la description atomique. C’est ce que traduit la phrase de Jean Perrin : « Expliquer du visible compliqué par de l’invisible simple ». Aujourd’hui, on est plus circonspect sur le fait de savoir ce qui est « simple » et ce qui est « compliqué ». Il reste toutefois que l’interprétation des phénomènes macroscopiques à l’aide de mécanismes microscopiques est l’une des aventures intellectuelles les plus fascinantes qui soit.
La simulation moléculaire donne une nouvelle dimension à l’étude de nombreux phénomènes physiques ou chimiques se produisant dans tous les états de la matière. Elle constitue une aide indéniable aux approches plus traditionnelles que sont la théorie analytique et l’expérience. Les approches théoriques font généralement appel à des approximations importantes et les interprétations des expériences sont souvent rendues délicates par la complexité des systèmes réels. De plus, il est pratiquement impossible de contrôler tous les paramètres expérimentaux ou de les faire varier sur une gamme aussi large qu’on le souhaiterait. Les comparaisons entre expériences et théorie peuvent être ardues, soit parce que l’observation expérimentale est difficile, soit parce que les modèles théoriques restent trop simples. Les simulations moléculaires se présentent comme une autre solution pour valider les modèles issus de la théorie ou pour en proposer d’autres. Les modèles utilisés doivent être suffisamment simples pour que les simulations soient réalisables en termes de temps de calcul, mais suffisamment réalistes pour reproduire au mieux la physico-chimie du phénomène étudié.
Mais l’intérêt de la simulation moléculaire va au-delà de simples validations des modèles théoriques et reproductions quantitatives des résultats expérimentaux. Elle peut se substituer à l’expérience et devenir prédictive lorsque celle-ci est irréalisable (conditions extrêmes de température et de pression, dangerosité des réactifs, coût trop élevé…). D’autre part, elle peut être une alternative à la théorie lorsque les modèles analytiques sont trop complexes ou inexistants. En outre, la simulation moléculaire permet une vision microscopique de la matière qui amène à mieux comprendre le déroulement de certains phénomènes (détail d’un mouvement moléculaire, d’une structure).
L'équipe "Théorie et Modélisation" du laboratoire TIM, par exemple, utilise des simulations de dynamique moléculaire à plusieurs échelles de représentation (ab initio, semi-empirique, classique, mixte QM/MM, gros grains et très gros grains) pour étudier une variété de systèmes. Ces simulations sont appliquées à des molécules en phase gazeuse, des liquides, des biomolécules immergées dans des liquides, des interfaces hétérogènes solide/liquide et air/liquide (incluant des électrolytes et des molécules organiques), ainsi qu'à des biomolécules traversant des nanopores. L'objectif est d'extraire des propriétés microscopiques et macroscopiques de la matière, telles que la structure, la dynamique, le transport, la réactivité chimique et les spectroscopies vibrationnelles.
Dans le domaine des interfaces aqueuses, des recherches fondamentales sur la caractérisation structurale, dynamique et spectroscopique d'interfaces complexes inhomogènes sont menées par des simulations de dynamique moléculaire ab initio (DFT-MD), souvent couplées à la spectroscopie non-linéaire SFG (Sum Frequency Generation). Ces travaux ont permis de définir des concepts clés comme les couches interfaciales BIL (Binding Interfacial Layer) et DL (Diffuse Layer), de révéler des réseaux de liaisons hydrogène, et de développer des descripteurs moléculaires universels d'hydrophilicité et d'hydrophobicité. De nouvelles espèces ont été identifiées à la surface de la silice en contact avec l'eau, et des modèles théoriques simplifiés pour le calcul de SFG ont été développés, ouvrant la voie à l'utilisation de l'IA pour prédire des structures d'interfaces aqueuses.
Le transport de biomolécules dans les nanopores est également étudié à l'aide de simulations de dynamique moléculaire, notamment avec le champ de force gros-grain MARTINI. Ces simulations permettent de comprendre les mécanismes moléculaires du transport à travers des nanopores protéiques comme l'alpha-hémolysine, reproduisant et expliquant des résultats expérimentaux sur l'asymétrie du courant et la sélectivité des ions, ainsi que sur la translocation de molécules d'ADN.
En chemo-informatique, l'équipe développe des méthodes basées sur la théorie des graphes pour analyser les systèmes moléculaires complexes, notamment la dynamique conformationnelle via les 2D-MolGraphs et le polymorphisme de graphes pour représenter les biomolécules à l'échelle des liaisons hydrogène. Des algorithmes de reconnaissance de motifs structuraux dans l'eau aux interfaces sont également développés, servant de base à des méthodes prédictives par intelligence artificielle. Des graphes vibrationnels sont utilisés pour la caractérisation des modes vibrationnels, et des Large Language Models (LLMs) sont explorés pour l'apprentissage de spectres infrarouges à partir de descripteurs moléculaires.
L'étude de la structure et de la dynamique d'espèces dans des fluides complexes aborde la stabilité d'assemblages atomiques ou moléculaires dans des environnements variés, des conditions cryogéniques aux régimes supercritiques. Des travaux récents incluent le calcul de propriétés thermodynamiques et de transport de solutions hydroalcooliques carbonatées, la discussion des processus de capillarité sur des corps célestes, et la mise en place de modèles mixtes classique-quantique pour la dynamique d'agrégats dans des nanogouttes d'hélium.
Enfin, l'étude des structures et spectroscopies de molécules en phase gazeuse, bien que moins prépondérante aujourd'hui, a vu l'application de simulations de dynamique moléculaire ab initio pour le calcul de spectroscopies, incluant les anharmonicités vibrationnelles et les effets de température. Ces recherches, menées en synergie avec des expériences, ont donné lieu à de nombreuses publications et collaborations internationales.
tags: #modelisation #thermodynamique #entre #molecules
