

La thermodynamique, science fondamentale régissant l'énergie et ses transformations, repose sur des principes qui guident notre compréhension des systèmes physiques et chimiques. Si le second principe, en se focalisant sur l'entropie, offre un critère d'évolution clair pour les systèmes thermiquement isolés, l'étude des transformations monothermes, c'est-à-dire celles se déroulant à température constante, requiert des outils adaptés. C'est dans ce contexte que les fonctions d'état telles que l'énergie libre (F) et l'enthalpie libre (G) prennent toute leur importance, agissant comme des potentiels thermodynamiques qui permettent de prédire le sens spontané des évolutions et d'atteindre l'équilibre.
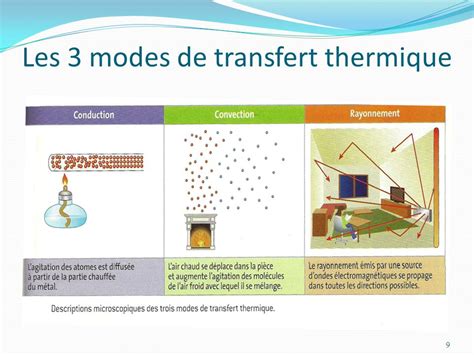
Avant de plonger dans les subtilités des potentiels thermodynamiques, il est essentiel de rappeler quelques notions fondamentales concernant les transferts thermiques et la thermochimie. Le transfert thermique, couramment appelé "chaleur" dans le langage quotidien, représente l'un des modes d'échange d'énergie thermique entre deux systèmes. Il se distingue du travail par sa nature microscopique et désordonnée. Trois modes principaux de transfert thermique coexistent : la conduction, la diffusion progressive de l'agitation thermique ; la convection, liée aux déplacements macroscopiques de matière ; et le rayonnement, propagation de photons. La quantité de chaleur, notée Q et mesurée en joules, représente l'énergie échangée par ces différents moyens. Une convention stipule que Q > 0 lorsque le système reçoit de l'énergie.
La thermochimie, quant à elle, se situe à l'interface entre la chimie et la thermodynamique. Elle étudie les phénomènes thermiques dans les milieux réactionnels. Les réactions chimiques peuvent être classées en deux catégories : exothermiques, qui dégagent de la chaleur, et endothermiques, qui en absorbent. La mesure des chaleurs de réaction s'effectue par calorimétrie, notamment à l'aide d'une bombe calorimétrique, inventée par Marcellin Berthelot, pionnier de cette discipline. L'application du second principe de la thermodynamique aux systèmes chimiques permet de prédire le sens des réactions et la position des équilibres.
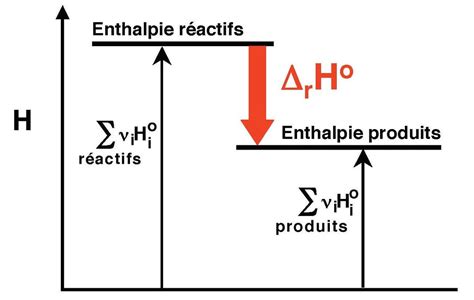
Au cours d'une réaction chimique, le système échange de l'énergie avec le milieu extérieur sous forme de transfert thermique. La quantité de chaleur échangée dépend des conditions expérimentales : à volume constant (transformation isochore), Qv = ΔU (variation d'énergie interne) ; à pression constante (transformation isobare), Qp = ΔH (variation d'enthalpie).
La thermodynamique s'articule autour de plusieurs principes fondamentaux, dont la compréhension est primordiale pour appréhender le comportement des systèmes.
Le principe zéro établit la notion d'équilibre thermique et fonde la thermométrie : si deux systèmes sont en équilibre thermique avec un troisième, ils le sont entre eux.
Le premier principe, également connu sous le nom de principe de conservation de l'énergie, stipule que l'énergie totale d'un système isolé reste constante. L'énergie ne peut être ni créée ni détruite, mais seulement transformée ou transférée. Cette loi, intimement liée à l'uniformité de la structure de l'espace-temps selon le théorème de Noether, se résume par le célèbre adage : "Rien ne se perd, rien ne se crée, tout se transforme."
Le second principe, ou principe d'évolution des systèmes, introduit la notion de dégradation de l'énergie et d'irréversibilité. L'énergie tend à passer de formes concentrées à des formes diffuses, comme la chaleur. Ce principe est intrinsèquement lié à l'entropie (S), une grandeur qui, pour un système isolé, ne peut qu'augmenter ou rester constante. Souvent interprétée comme une mesure du désordre, l'entropie de Shannon, issue de la théorie de l'information, offre une perspective statistique sur ce concept. À la différence du premier principe, le second principe a une origine essentiellement statistique.
Le troisième principe concerne l'entropie à l'approche du zéro absolu de température. Il stipule que l'entropie d'un système en équilibre interne tend vers une constante universelle (souvent prise comme nulle par convention) lorsque la température s'approche de zéro Kelvin. Ce principe est lié à l'état quantique fondamental d'un système.
Au-delà de ces principes, la thermodynamique s'appuie sur des concepts tels que les variables d'état (P, T, V, U, H, S, F, G), qui décrivent l'état d'un système, et les transformations thermodynamiques, qui sont les évolutions d'un système d'un état initial à un état final. Les variables peuvent être extensives (proportionnelles à la quantité de matière, comme la masse ou le volume) ou intensives (indépendantes de la quantité de matière, comme la température ou la pression). Une transformation quasistatique est une transformation suffisamment lente pour que le système puisse être considéré comme passant par une succession d'états d'équilibre.
Dans le cadre des transformations monothermes (à température constante) et sans travail reçu autre que celui des forces de pression, l'évolution spontanée d'un système est gouvernée par la diminution de certaines fonctions d'état.
Pour les transformations monothermes et isochores (volume constant), sans travail reçu autre que celui des forces de pression, l'énergie libre de Helmholtz, notée F, agit comme potentiel thermodynamique. Elle est définie par la relation :
$$ F = U - TS $$
où $U$ est l'énergie interne, $T$ la température absolue et $S$ l'entropie.
Lors d'une transformation monotherme, le travail récupérable ($-\text{W}$) est inférieur ou égal à la chute de F. L'égalité n'est atteinte que pour une transformation réversible. Autrement dit, F diminue lors de l'évolution spontanée du système et atteint son minimum à l'équilibre thermodynamique. Les variables naturelles de F sont la température (T) et le volume (V), ce qui conduit à l'identité thermodynamique :
$$ dF = -SdT - pdV $$
F est une fonction caractéristique, permettant de décrire complètement le comportement thermodynamique du système à l'équilibre.
Pour les transformations monothermes et isobares (pression constante), sans travail reçu autre que celui des forces de pression, l'enthalpie libre de Gibbs, notée G, est le potentiel thermodynamique pertinent. Elle est définie par :
$$ G = H - TS $$
où $H$ est l'enthalpie ($H = U + pV$).
Lors d'une transformation monotherme et isobare, le travail récupérable autre que celui des forces de pression ($-\text{W}'$) est inférieur ou égal à la chute de G. L'égalité est vérifiée pour une transformation réversible. Ainsi, G diminue lors de l'évolution spontanée du système et est minimale à l'équilibre thermodynamique. Les variables naturelles de G sont la température (T) et la pression (p), et son identité thermodynamique s'écrit :
$$ dG = -SdT + Vdp $$
Comme F, G est une fonction caractéristique du système thermodynamique. L'enthalpie libre est particulièrement utile en chimie pour étudier les équilibres chimiques à pression et température constantes. Le signe de la variation de l'enthalpie libre de réaction ($\Delta G$) indique le sens d'évolution de l'équilibre : si $\Delta G < 0$, la réaction progresse spontanément dans le sens direct ; si $\Delta G > 0$, elle progresse dans le sens inverse ; si $\Delta G = 0$, le système est à l'équilibre.
Les potentiels thermodynamiques, F et G, trouvent des applications majeures dans divers domaines, notamment en chimie pour prévoir le sens des réactions et la position des équilibres, mais aussi dans l'étude des changements d'état.
Les changements d'état, tels que la fusion, la vaporisation ou la sublimation, sont des transitions de phase du premier ordre. Ils s'accompagnent d'une absorption ou d'un dégagement de chaleur (chaleur latente) et d'une variation d'entropie. Les tables thermodynamiques fournissent les valeurs de ces chaleurs latentes, essentielles pour les calculs d'ingénierie.
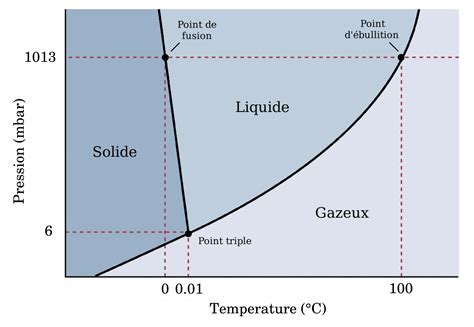
Les diagrammes de phases, comme les diagrammes $(P,T)$ ou $(p,v)$, représentent graphiquement les différentes phases d'une substance et les conditions sous lesquelles elles coexistent. Le point triple est un point unique où les trois phases (solide, liquide, gaz) coexistent en équilibre. Au-delà du point critique, il n'y a plus de distinction nette entre phase liquide et gazeuse.
La règle des moments, appliquée aux diagrammes $(p,v)$ par exemple, permet de déterminer la composition d'un mélange diphasé connaissant le titre massique ou molaire.
Les machines thermiques, qui transforment l'énergie thermique en travail, sont au cœur de nombreuses applications industrielles. Le cycle de Carnot, composé de quatre transformations réversibles (deux isothermes et deux adiabatiques), définit le rendement maximal théorique qu'une machine thermique peut atteindre entre deux sources de chaleur. Le rendement d'une machine thermique est défini comme le rapport entre le travail utile produit et la chaleur absorbée. Pour les machines frigorifiques et les pompes à chaleur, on utilise l'efficacité (COP), qui mesure le rapport entre le transfert thermique utile et le travail consommé.
Les fonctions d'énergie libre de Helmholtz (F) et d'enthalpie libre de Gibbs (G) sont des outils indispensables en thermodynamique pour analyser les transformations monothermes. Elles agissent comme des potentiels qui guident l'évolution spontanée des systèmes vers un état d'équilibre, en particulier dans les processus chimiques et les changements d'état. Leur compréhension approfondie permet de modéliser et de prédire le comportement de systèmes complexes dans diverses conditions, ouvrant la voie à des applications technologiques et scientifiques avancées. L'étude de ces potentiels, couplée aux principes fondamentaux de la thermodynamique, offre une clé de lecture essentielle pour appréhender les phénomènes énergétiques qui régissent notre univers.
tags: #cas #thermodynamique #possible #schema
